

Joseph R, Anticaglia, MD
Medical Advisory Board
Sickle cell disease (SCD) is an inherited blood disease that’s passed down from parent to child. This genetic disorder causes the body to make abnormal red blood cells (RBC’s). Hemoglobin is the protein in the RBC’s that delivers oxygen to all parts of the body and sends carbon dioxide to the lungs to be exhaled from the body.
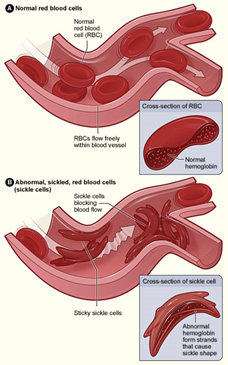
Red blood cells normally make hemoglobin A. These cells are round, smooth, soft enabling hemoglobin to carry oxygen easily throughout the circulatory system. In sickle cell disease, the RBC’s make the deformed protein hemoglobin S (HgbS). The abnormal HgbS cells have been described as being crescent or sickled shaped, pointed, hard and sticky.
When passing through small blood vessels, sticky sickle cells get stuck together and often completely block the tiny vessels. Hemoglobin S cells can also damage larger blood vessels by clinging to their walls causing them to scar and narrow, and at times, block them.
HgbS red blood cells are fragile, rupture easily (hemolysis) leading to anemia. They have a short life span surviving just 10 to 20 days compared to 120 days of normal RBC’s. The combination of blood vessel blockage, anemia and the buildup of hemoglobin in the body cause symptoms beyond excruciating painful crises.
Signs and Symptoms
The symptoms vary from person to person. They may be mild or severe. They include:
Anemia is the most common finding. Because of the anemia, people complain of being tired, lightheaded or dizzy. They let their doctor know that at times they feel out of breath and feel as if the heart is racing. Paleness may involve the lips and nail beds. Viral or bacterial infections can provoke life threatening hemolytic anemia causing the rupture of red blood cells
Painful Crises: When sickle cells block blood vessels, they cut off the oxygen supply to the area and cause pain and other symptoms. Dehydration, infections or stress can trigger recurrent, painful episodes in any part of the body but most often in the arms, legs and chest. These episodes can last from a few hours to several days.
Jaundice: Yellowing of the skin, whites of the eyes and mouth (jaundice) is a common symptom of people with SCD. This happens because fragile sickle cells break down faster than the liver can filter them out. Bilirubin builds up causes the whites of the eyes and skin to become yellow. Sickle cells rupture every 10 to 20 days.
Chest Pain: Persons with SCD need emergency care if they have acute chest pain. This takes place when sticky sickle cells block the small blood vessels of the lungs cutting of their oxygen supply. Apart from chest pain, they may have fever and severe cough.
Stroke: Sickle cells cause problems whenever they block blood vessels and interrupt the supply of oxygen to tissues and organs. A stroke and brain damage are the consequences when the blockage occurs in the brain.
Spleen Healthy blood cells “pass go” and move through the spleen to circulate throughout your bloodstream. Damaged or malformed blood cells) get stopped by the spleen’s filter system and get broken down. When sickle cells get stuck in the spleen, the spleen enlarges (splenomegaly) and there could be a dangerous drop in hemoglobin circulating through your body.
Priapism is a symptom among patients with SCD. Blood vessels of the penis get blocked causing great pain. Repeated episodes can lead to impotence
What Causes Sickle Disease?
A defected gene causes sickle cell disease. Children who make hemoglobin S in their red blood cells got the sickle cell gene from one or both of their parents. If both parents pass the sickle cell gene onto their child, the child will inherit sickle cell disease (SS). When a child receives the sickle cell gene from one parent, that child will inherit sickle cell trait (S) and not the symptoms of sickle cell disease.
If one parent has sickle cell disease and one parent has sickle cell trait, there is a 50% chance that their children will be born with sickle cell disease.
Diagnosis
A special blood test is performed to determine if a child has sickle cell disease or trait. Parents may decide to have this test before they plan to have children. In the United States, this blood test is done as part of routine newborn screening=ng. Doctors can diagnose SCD by taking and testing a sample of the amniotic fluid that surrounds the fetus before it’s born.
Treatment
According to NIH, “There are treatments that can help relieve symptoms, lessen complications, and prolong life:
- Antibiotics to try to prevent infections in younger children
- Pain relievers for acute or chronic pain
- Hydroxyurea, a medicine that has been shown to reduce or prevent several SCD complications. It increases the amount of fetal hemoglobin in the blood. This medicine is not right for everyone; talk to your health care provider about whether you should take it. This medicine is not safe during pregnancy.
- Childhood vaccinations to prevent infections
- Blood transfusions for severe anemia. If you have had some serious complications, such as a stroke, you may have transfusions to prevent more complications.
The only cure for SCD is bone marrow or stem cell transplantation. Because these transplants are risky and can have serious side effects, they are usually only used in children with severe SCD.”
CRISPR-Cas9
Scientists are hopeful of using CRISPR-Cas9 to treat and cure genetic flaws in children and adults afflicted with SCD. This technology, gene editing or genetic engineering, makes use of guide proteins (gRNA) to locate abnormal genes and cutting proteins (Cas9) to remove or repair flawed genes or insert healthy genes into a section(s) of the genome+ (see doctor column reference below).
Individuals with SCD face troublesome, lifelong challenges including painful crises, stroke and a median life expectancy between 40 to fifty years of age. They can encounter injury to the liver, kidney or spleen and visual problems due to damage to the retina similar to diabetic patients
By utilizing CRISPR-Cas9, there’s hope that in the not too distant future there will be a cure for sickle cell disease that will be more reliable, safer, faster and cheaper. As with any new technology, scientists urge caution because of unforeseen complications.
Reference
- Haydar Frangoul, M. D et al; CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia; The N. Eng. J. Med.; January 21, 2021
- NIH; Sickle Cell Disease
- Lloyd E. Damon; Charalambos Andreadis; Blood Disorders; Current Med Dx & Rx. Lamge.2018
- Joseph R. Anticaglia, MD; A 21st Century Scientific Revolution? CRISPR-Case 9; Doctor’s Column, 2021
- *Reach out to SCD centers if available, to obtain the latest information to manage your disease.
This article is intended solely as a learning experience. Please consult your physician for diagnostic and treatment options.